Open Access | Commentary
This work is licensed under a Creative
Commons Attribution-ShareAlike 4.0 International License.
Targeting IGF1R signaling for brain aging and Alzheimer’s disease
* Corresponding author: Warren Ladiges
Mailing address: Department of Comparative Medicine, School
of Medicine, University of Washington, Seattle, WA, USA.
Email: wladiges@uw.edu
Received: 20 December 2022 / Accepted: 22 December 2022 / Published: 29 December 2022
DOI: 10.31491/APT.2022.12.103
Abstract
The role of IGF1R signaling in the brain and its relationship to aging and neurological dysfunction is controversial. Because it was shown that low IGF1R activity consistently improved myocardial bioenergetics and function in hearts from aging mice, but not hearts from young mice, it was of interest to investigate this relationship in brain aging. We used CRISPR technology to develop a mouse model with targeted replacement of mouse IGF1R with the equivalent of the human R407H (IGF1RR407H) variant enriched in centenarians with a reduction in IGF1R protein activity. Middle-aged mice show improved cognitive performance thus possibly modeling IGF1R signaling in the aging brain, similar to what was reported in the aging heart. Because Alzheimer’s disease (AD) is an age-related disease, specific IGF1RR407H pathways could be therapeutic targets in mice with AAV vector-based AD as well as for overall brain aging.
Keywords
IGF1R signaling, IGF1RR407H variant, brain aging, cognition, Alzheimer’s disease
Signaling of insulin/insulin-like growth factor-1 (IGF1)
through the IGF1-receptor (IGF1R) is central to processes
that modulate aging and lifespan in model organisms [1-2],
as well as mortality and morbidity in humans [3]. IGF1R
signaling controls cellular growth and metabolism through
a wide variety of downstream pathways. A recent study
by Abdellatif et al. [4] showed that low IGF1R activity
consistently improved myocardial bioenergetics and function in hearts from aging mice, but not hearts from young
mice, in an autophagy-dependent manner. They concluded
that the relationship between IGF1R signaling and cardiac health is not linear, but biphasic, since mice with low
IGF1R signaling exhibited poor cardiac function early in
life, while young mice with increased IGF1R signaling
showed a superior cardiac function that progressively declined with increasing age. Moreover, analyses of serum
IGF1 levels and various clinical diseases lend further
support for such antagonistic pleiotropy [5]: protection
of young individuals with high IGF1 levels and increased
disease risk in older individuals with higher IGF1 levels.
While much work on IGF1R has focused on peripheral
organs, there is reason to assume the same mechanism occurs in the brain. It has been shown that increased IGF1R
signaling was apparent in postmortem brains of Alzheimer’s disease (AD) patients [6], suggesting that a long-term
increase in activity is associated with the progression of
AD neuropathology. Genetic ablation of IGF1R in neurons of aging mice can protect against neuroinflammation
and memory impairment induced by A beta oligomers [7].
However, the role of IGF1R signaling in the brain and its
relationship to aging and neurological dysfunction is controversial. For example, Tazearslan et al. [8] reported that
age-related astrocyte dysfunction caused by diminished
IGF1R signaling may contribute to the pathogenesis of
AD and other age-related cognitive impairments. A possible explanation for the opposite effects to be in play may
be that different downstream pathways respond differently
with increasing age. Evidence to support this possibility
showed that Ashkenazi Jewish centenarians were found to
be enriched with variants in the IGF1R gene [9]. Individuals carrying two specific variants, A37T and R407H, had a
significant reduction in IGF1R protein and its phosphorylation levels.
Based on these observations, we used CRISPR technology
to develop a mouse model with a targeted replacement of
mouse IGF1R with the equivalent of the human R407H
(IGF1RR407H) variant [10]. The objective was to establish a precision animal model that could help provide definitive
answers to the role of IGF1R signaling in aging, including
age-associated conditions such as cardiac aging and neurodegeneration. Since IGF1RR407H disrupts phosphorylation levels of ERK and AKT but not IGF1R-IGF1 binding, these specific pathways (and probably others) can be
interrogated using this newly developed mouse line. Thus,
we can study how a single variant may down-regulate IGF1R signaling in a more precise pathway-specific manner
to provide a protective environment to delay brain aging
and prevent the development of neurological conditions
such as Alzheimer’s disease (AD). Is this mouse line then
a model of the biphasic signaling by IGF1R in the aging
brain similar to what Abdellatif et al. [4] reported in the
aging heart? Perhaps, but more work is needed. We have
not observed any adverse phenotypic effects at younger
ages but do see an improved cognitive performance at
older ages compared to wild-type littermates.
These published and unpublished observations provide
the rationale to develop therapeutic targets for the IGF1RR407H variant to prevent or delay brain aging. There
is support for IGF1R as a target for therapeutic intervention of aging. Huffman et al. [11] showed that 18-monthold female but not male mice treated with a monoclonal
antibody (mAb) targeting IGF1R (L2-Cmu, Amgen Inc)
had improved health span and increased median lifespan
by 9 percent along with a reduction in neoplasms and
inflammation compared to controls. These effects were
achieved at advanced ages, suggesting that IGF1R mAbs
could represent a promising therapeutic candidate to delay
aging. In a second study, picropodophyllin (PDP), a selective, competitive, and reversible inhibitor of IGF1R that is
brain penetrant, was given to a mouse model of AD amyloid neuropathology (APP/PS1 Tg mice) daily for 7 days
IP at 1 mg/kg/day [12]. PDP attenuated levels of Aβ40
and 42 and decreased microgliosis and p-tyrosine in the
hippocampus.
Since AD is an age-related disease, drugs that prevent or
slow down brain aging should also be effective in helping resist the development of AD neuropathology and the
associated severe cognitive impairment. So far, there has
been no single therapeutic compound that has been shown
in clinical trials to have significant beneficial reversible effects even though preclinical animal studies were
promising. There may be a disconnect between currently
available mutant FAD mouse models and the ability to
predict clinical outcomes. In most of the transgenic lines,
a significant increase in APP production begins early in
life possibly in utero, which may trigger consequences
that alter aging and the rate of aging, and likely does not
mimic the biochemical changes observed in AD. A further
complication is the lack of the mixed amyloid and Tau
pathology that characterizes AD. We have developed an
aging mouse model of AD neuropathology using an AAVbased transfer of Aβ42 and mutant MAPT (tau P301L)
[13], which allows us to test drugs in a more realistic preclinical manner. Middle-aged and older aged mice can be
obtained from the United States National Institute on Aging Aged Rodent Colony and used in drug testing, without having to wait several years for young mice to age.
Drugs can be tested in older mice over several months so
different therapeutic strategies can readily be carried out.
However, a high throughput drug screening system for
IGF1RR407H or other IGF1R variants has yet to be developed so candidate drugs are somewhat limited. RNA sequencing could help determine whether to target specific
downstream pathways. Regardless, there is a rationale for
identifying drugs that target IGF1R signaling based on the
beneficial aspects of significant reductions in activity at
older ages. A preclinical mouse model is in place to speed
up the process of moving from preclinical testing to clinical trials for treating and preventing AD (Figure 1).
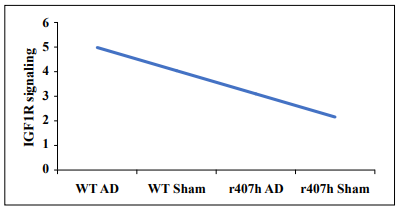
Figure 1. A hypothetical example of the effect of the IGF1RR407H variant on IGF1R signaling on a scale of 1 to 5 (low to high) in 22-month-old C57BL/6 mice with or without AAV vector-transferred Alzheimer's disease. Abbreviations: AD = AAV Aβ42+tauP301L; r407h = the IGF1R variant; Sham = AAV vector only; WT = wild-type littermates.
Declarations
Authors’ contributions
All authors contributed to the writing of this manuscript.
Availability of data and materials
Not applicable.
Financial support and sponsorship
Supported in part by NIH grants R01 AG067193 (Ladiges and Darvas, coPI’s) and R01 AG057381 (Ladiges, PI).
Conflicts of interest
Warren Ladiges and Martin Darvas are members Editorial Board of Aging Pathobiology and Therapeutics. The authors declare that there are no conflicts and and were not involved in the journal’s review or decisions related to this manuscript.
References
1. Kenyon CJ. The genetics of ageing. Erratum in: Nature, 2010, 464(7288): 504-512. [Crossref]
2. Milman S, & Barzilai N. Dissecting the Mechanisms Underlying Unusually Successful Human Health Span and Life Span. Cold Spring Harb Perspect Med, 2015, 6(1): a025098. [Crossref]
3. Zhang WB, Aleksic S, Gao T, Weiss EF, Demetriou E, Verghese J, et al. Insulin-like Growth Factor-1 and IGF Binding Proteins Predict All-Cause Mortality and Morbidity in Older Adults. Cells, 2020, 9(6). [Crossref]
4. Abdellatif M, Madeo F, Kroemer G, & Sedej S. Spermidine overrides INSR (insulin receptor)-IGF1R (insulinlike growth factor 1 receptor)-mediated inhibition of autophagy in the aging heart. Autophagy, 2022, 18(10): 2500-2502. [Crossref]
5. Zhang WB, Ye K, Barzilai N, & Milman S. The antagonistic pleiotropy of insulin-like growth factor 1. Aging Cell, 2021, 20(9): e13443. [Crossref]
6. Zhao Y, Xie YZ, & Liu YS. Accelerated aging-related transcriptome alterations in neurovascular unit cells in the brain of Alzheimer’s disease. Front Aging Neurosci, 2022, 14: 949074. [Crossref]
7. George C, Gontier G, Lacube P, François JC, Holzenberger M, & Aïd S. The Alzheimer’s disease transcriptome mimics the neuroprotective signature of IGF-1 receptordeficient neurons. Brain, 2017, 140(7): 2012-2027. [Crossref]
8. Logan S, Pharaoh GA, Marlin MC, Masser DR, Matsuzaki S, Wronowski B et al. Insulin-like growth factor receptor signaling regulates working memory, mitochondrial metabolism, and amyloid-β uptake in astrocytes. Mol Metab, 2018, 9: 141-155. [Crossref]
9. Tazearslan C, Huang J, Barzilai N, & Suh Y. Impaired IGF1R signaling in cells expressing longevity-associated human IGF1R alleles. Aging Cell, 2011, 10(3): 551-554. [Crossref]
10. Dou Y, Darvas M, Sharma K, Mathieu J, John Morton J, Tan H, et al. Development of an IGF1R longevity variant mouse line using CRISPR/Cas9 genome editing. J Tre Bio Res, 2020.[Crossref]
11. Mao K, Quipildor GF, Tabrizian T, Novaj A, Guan F, Walters RO, et al. Late-life targeting of the IGF-1 receptor improves healthspan and lifespan in female mice. Nat Commun, 2018, 9(1): 2394. [Crossref]
12. Sohrabi M, Floden AM, Manocha GD, Klug MG, & Combs CK. IGF-1R Inhibitor Ameliorates Neuroinflammation in an Alzheimer’s Disease Transgenic Mouse Model. Front Cell Neurosci, 2020, 14: 200. [Crossref]
13. Darvas M, Keene D, & Ladiges W. A geroscience mouse model for Alzheimer’s disease. Pathobiol Aging Age Relat Dis, 2019, 9(1): 1616994. [Crossref]