Open Access | Review
This work is licensed under a Creative
Commons Attribution-ShareAlike 4.0 International License.
Nrf2 participates in progression of osteoarthritis through modulating redox balance
# Zihan Deng and Chuan Yang contributed equally to this work.
* Corresponding author: Yueqi Chen
Mailing address: Department of Orthopedics, Southwest Hospital, Third Military Medical University (Army Medical University), Chongqing, China.
Email: chenyueqi1012@sina.com
This article belongs to the Special Issue: Skeletal Aging; Cellular and Molecular Mechanisms
Received: 06 March 2023 / Revised: 25 May 2023 / Accepted: 14 June 2023 / Published: 28 June 2023
DOI: 10.31491/APT.2023.06.112
Abstract
Osteoarthritis (OA) is one of the most common degenerative joint diseases associated with aging, obesity and joint trauma, and is always associated with pain, joint deformity and dysfunction. Recent studies have shown that OA is closely related to oxidative stress, which serves as a major cause of chronic inflammation in cartilage, leading to irreversible structural changes in the joint. As a transcription factor, nuclear factor erythroid 2-related factor 2 (Nrf2) plays an important role in the antioxidant system, which regulates the expression of cytoprotective genes to facilitate the elimination of reactive oxygen species (ROS). In this review, we have summarized the dramatic function of oxidative stress in OA pathology, established a complex regulatory network of Nrf2 in OA progression, and aimed to provide new insights into the treatment of OA.
Keywords
Osteoarthritis, nuclear factor-erythroid 2-related factor, oxidative stress, redox balance, chondrocyte
Introduction
As a common chronic degenerative disease, osteoarthritis (OA) is associated with increasing obesity and global
population aging, and has become a major public health
burden, currently affecting more than 500 million people
worldwide [1]. OA is mainly characterized by pain as the
primary clinical manifestation, which gradually affects the
stability of joint motion and ultimately leads to the disability, thereby reducing the quality of patient’s life. To
date, the risk factors that contribute to the development of
OA are diverse, including aging, trauma, obesity, as well
as mechanical loading and many other factors [2]. Aging
is one of the most evident risk factors for the pathogenesis of OA, and studies have shown the presence of a
large number of senescent chondrocytes in the cartilage
of hip and knee joints in OA patients [3, 4]. Specifically,
with the progression of aging, senescent chondrocytes
gradually accumulate in OA joints and secrete a variety
of senescence-associated secretory phenotypes (SASPs),
including various pro-inflammatory cytokines and matrix
metalloproteinases (MMPs), which can activate chronic
inflammatory responses and induce oxidative stress, thus
contributing to the accumulation of reactive oxygen species (ROS) and leading to the disruption of antioxidant
enzymes and ROS scavenging systems [5]. ROS are the
toxic by-products of aerobic metabolism that are both essential and lethal for cell behavior [6]. On the one hand,
ROS are required for cellular signal transduction in many
fundamental biological processes, including cell growth
and differentiation [7]. On the other hand, ROS can be
toxic to DNA, proteins and lipids, leading to severe oxidative damage to cells and even cell death. Recent studies
have demonstrated that low-grade chronic inflammation
increases ROS accumulation and leads to excessive oxidative stress, which causes abnormal cell metabolism in
chondrocytes, thereby accelerating cartilage degradation
[8]. Therefore, maintaining oxidant-antioxidant balance
and redox homeostasis in OA chondrocytes is essential for OA treatment and remission.
Nuclear factor-erythroid 2-related factor (Nrf2) is widely
recognized as an important component of the antioxidative stress system, which is heavily involved in the cellular defense against multiple pathological stressors and
maintains the homeostasis of the intracellular environment. Nrf2 functions cytoprotective roles by modulating
the expression of various important genes involved in the
scavenging of ROS, reactive nitrogen species (RNS) and
electrophiles. Specifically, Nrf2-induced antioxidation is
generally achieved by enzymes that regulate the biological
synthesis, utilization, and regeneration of the reduced glutathione (GSH). In the process of GSH synthesis, Nrf2 targets three key enzyme genes, including glutamate cysteine
ligase catalytic (GCLC), modulator (GCLM) subunits,
and glutathione synthetase (GSS). In addition, the redox
cycling enzymes thioredoxin, thioredoxin reductase and
many other glutathione S-transferases that are responsible
for scavenging ROS are also the targets of Nrf2 [9]. Interestingly, Nrf2 also forms a large network of cooperating
enzymes in the regulation of basal metabolic processes,
mainly including metabolic enzymes involved in the pentose phosphate pathway and fatty acid metabolism, which
form a potential bridge between redox and intermediary
metabolism [5, 10-12].
It has been well studied that the Nrf2 protein consists of
seven functional domains, including Neh2, Neh4, Neh5,
Neh7, Neh6, Neh1, and Neh3 from the N-terminus to the
C-terminus. Among these functional domains, Neh2 and
Neh6 affect Nrf2 stability, with the Neh2 domain responsible for Keap1-mediated Nrf2 degradation, and Neh6 involved in the negative modulation of Nrf2 via the serinerich domain [13]. Neh3 could recruit the chromo-ATPase/
helicase DNA-binding protein, and Neh4 and Neh5 form
the transactivation domain to recruit CREB-binding protein (CBP), which participates in Nrf2-dependent transcriptional activation of genes with the ARE sequence
in the promoter [14]. The Neh1 domain promotes Nrf2
heterodimerization with small musculoaponeurotic fibrosarcoma (Maf) proteins to achieve DNA binding [15, 16].
In addition, Neh1 interacts with the ubiquitin-conjugating
enzyme UbcM2, which enhances the transcriptional activity of endogenous of Nrf2 [17]. The Neh7 domain binds
and interacts with the retinoid X receptor alpha (RXRα),
which is a repressor of Nrf2 [18]. Recent studies have
demonstrated that Nrf2 plays a critical role in numerous
common human diseases associated with oxidative stress,
such as type 2 diabetes and cardiovascular disease. In fact,
Hinoi et al. and Solomon et al. found that appropriate
Nrf2 activity is also essential for chondrocyte differentiation and maturation and cartilage metabolism [19, 20]. In
addition, Nrf2 was also found to modulate osteoclastogenesis in bone resorption and remodeling, further demonstrating the key role of Nrf2 in bone homeostasis [21].
Therefore, we summarized the underlying mechanisms
by which Nrf2 regulates oxidative stress in OA and aimed
to develop novel biopharmaceuticals for the treatment of
OA.
Pathogenesis of OA
OA, the most common chronic joint disease with increasing incidence in an aging and increasingly obese population, is a major cause of disability in the elderly and has
a significant impact on socioeconomic costs [22-24]. In
recent years, researchers have increasingly focused on
the important role of articular cartilage in the progression
of OA. Articular cartilage, a specialized dense connective tissue, is composed of chondrocytes and extracellular
matrix, and has avascular and alymphatic properties that
affect its natural ability to self-repair [22, 23]. Articular
cartilage covers the load-bearing surface of the bone to
form a smooth articular contact surface that absorbs external mechanical pressures or stimuli to achieve painless
joint motion [24].
OA is a degenerative change that disrupts the dynamic
balance between joint tissue destruction and repair, resulting in the loss of normal physiological joint function [25].
The complex pathogenesis of this disease involves several
aspects, such as increased inflammatory components,
mechanical overload, and metabolic disorders, which
gradually contribute to structural changes in the articular
and periarticular tissues, specifically leading to apoptosis
of chondrocytes and progressive degeneration of articular
cartilage [22, 26-29]. Chondrocytes in OA are activated
by various factors that lead to cartilage degradation, such
as high mechanical stress, erosion of a large number of
pro-inflammatory cytokines, imbalanced oxidative stress,
and senescence, among others, leading to the disruption
of joint homeostasis and further inducing the production
of matrix-degrading enzymes, resulting in bone metabolic
disorders and aberrant inflammatory osteolysis [30-32].
Initially, alterations in chondrocyte function in OA cause
progressive cartilage degradation and destruction, which
may be accompanied by persistent secondary inflammation [33]. As cartilage degeneration progresses, osteoclastmediated bone resorption is abnormally accelerated,
leading to bone cysts and sclerotic bone formation. In
addition, cartilage and bone loss disrupts bone matrix homeostasis and triggers compensatory osteoblast-mediated
bone remodeling. In advanced disease, deeper cartilage
fissures are followed by osteoblast oversynthesis, leading
to osteophyte formation at the joint margins, expansion of
the calcified cartilage zone, and periarticular fibrosis [22,
34].
It is noteworthy that hypertrophic chondrocytes exhibit
increased synthetic activity during the repair process, producing numerous pro-inflammatory mediators and stromal
degradation products that act on the adjacent synovium
to facilitate proliferation and inflammatory responses,
accompanied by tissue hypertrophy and angiogenesis
[22, 35]. Insights into the pathophysiology of the disease
indicate that mutations or errors in the gene expression
of matrix molecules and certain factors that modulate
the synthesis of matrix components may lead to chondrocyte hypertrophy and dysfunction, resulting in chondrodysplasia at a relatively early age [34]. Furthermore,
chondrocytes located near load-bearing regions are more
likely to acquire this altered phenotype, and susceptibility to OA also increases with age. In the middle and late
phases of OA, various cell types (including chondrocytes, osteoclasts, osteoblasts, and immune cells, etc.) may be
involved in the pathogenesis, and all of them may exhibit
abnormal gene expression and disruption of the oxidoantioxidant balance, ultimately leading to OA predisposition in the elderly population [5].
Dramatic role of oxidative stress in OA pathology
Over the past few decades, numerous studies have demonstrated that oxidative stress plays an integral role in the
pathogenesis of several age-related diseases, including
cardiovascular, bone, renal, and neurodegenerative diseases [36]. Furthermore, increased oxidative stress and
decreased mitochondrial antioxidant capacity affect physiological cellular signaling pathways, which may contribute to senescence through progressive loss of cellular
integrity and disruption of tissue homeostasis [37, 38]. In
the context of OA, there is a growing consensus that oxidative stress is a driver of an imbalance between catabolic
and anabolic signals in cartilage, which progressively induces bone matrix degradation as the disease progresses,
leading to aberrant inflammatory osteolysis [39].
ROS, consisting of superoxide anion (O2·−), hydrogen
peroxide (H2O2), and hydroxyl radical (OH), among others, are by-products of normal cellular metabolism that are
generated in electron transport chain reactions and are primarily responsible for transferring electrons to molecular
oxygen in the mitochondria [40]. This process is limited
by the oxidoreductase p66Shc, which translocates to mitochondria in response to exogenous signals such as growth
factor deprivation, oxidative stress, and ultraviolet radiation [40, 41]. ROS in mitochondria can cause oxidative
stress and is a predominant regulator of cellular senescence, inducing multiple genes to facilitate mitochondrial
dysfunction, swelling and apoptosis associated with aging,
as well as triggering senescence or dedifferentiation of
chondrocytes [42]. Additionally, some ROS are produced
by non-mitochondrial pathways, namely NADPH oxidase
(NOX) or dual oxidase (DUOX), which exist in discrete
regions of plasma or endosomal membranes. And NOX
enzymes can regulate downstream signals for cell activation, differentiation, proliferation, and apoptosis in healthy
cells, whereas they are responsible for confining H2O2 to
specific cellular microdomains and preventing its diffusion
into the cytoplasm, thereby blocking abnormal signaling
[43, 44]. It has been reported that ROS (such as H2O2 and
O2·−) and RNS [including ·NO and peroxynitrite (ONOO-)] play an important role in regulating chondrocyte function, disrupting cartilage homeostasis, and inducing the
progression of osteoarthritis [45-47].
In chondrocytes, osteoblasts, and osteoclasts of OA, aberrant ROS signaling is often accompanied by a spatiotemporal progression of damage from the articular surface
to the subchondral bone. Yudoh et al. found that the antioxidant capacity in the degenerated cartilage region of
OA patients was dramatically lower than that in the intact
cartilage region, indicating that oxidative damage was
increased in degenerated cartilage compared with normal
cartilage [48]. Interestingly, based on in vitro experiments,
H2O2-cultured chondrocytes had shorter telomere length.
In addition, studies have shown that when OA cartilage
tissue is treated with H2O2, glycosaminoglycans (GAGs),
a long linear polysaccharide that can attach to the articular
surface with lubricating and protective effects, are gradually reduced in a time-dependent manner. Nevertheless,
the use of antioxidants has been confirmed to reverse the
above effects, limiting the loss of GAGs and maintaining telomere length [49]. In fact, ROS exert a significant
effect on the dynamic balance of osteoclast-mediated
bone resorption and osteoblast-mediated bone remodeling under physiological conditions, which is conducive to
maintaining bone integrity. However, abnormal levels of
ROS can negatively regulate mitochondrial function and
lead to changes in signal transduction pathways and gene
expression, which may induce chondrocyte apoptosis and
senescence, ultimately contributing to cartilage degeneration, as well as alterations in subchondral bone and bone
remodeling processes [5].
It is well known that ROS production and clearance in
cells are in a state of dynamic equilibrium under physiological conditions, thus maintaining the homeostasis of the
internal environment in the cytoplasm. One caveat is that
there are some scavenging systems that can be used to detoxify ROS, consisting of catalase, SOD, as well as GSH
peroxidase and reductase, etc [45]. As a dimeric cytosolic
enzyme, SOD1 binds copper and zinc (Cu/Zn-SOD),
while SOD2 is a mitochondrial homotetramer binding one
manganese ion per subunit (Mn-SOD). Both enzymes are
responsible for the conversion of superoxide to H2O2 and
diatomic oxygen. And catalase, composed of a tetrameric
protein, is able to convert H2O2 to H2O and gaseous O2.
Moreover, in the cytosol, the GSH peroxidase and GSH
reductase system maintains the reducing environment
in cells. When ROS production escapes the antioxidant
systems and mechanisms, cells are adversely affected by
oxidative stress and become susceptible to activation of
apoptotic pathways. ROS-mediated damage can often be
reversed by repair, replacement, degradation, or sequestration of the damaged macromolecules, but in some cases
the stress can be sustained, driving mitochondrial and cell
death or mutagenesis [40, 41]. The biological effects of
ROS in all cell types are due to hyperperoxidation, protein
carbonylation, direct DNA damage, telomere shortening,
epigenetic changes in gene expression and failure of DNA
repair, alterations in receptor and metabolic pathways, and
autophagy. However, the sources of ROS may differ in
different cell types and may depend on the functional and
metabolic state of the cell type.
Nrf2 and redox balance during OA progression
Aging is known to be an important pathogenic factor in
OA, and Nrf2 may play an indispensable role in inhibiting
cellular aging through the antioxidant system. Research
has shown that the activity of Nrf2 gradually decreases during the aging process of human fibroblasts. As expected, silencing of Nrf2 is able to induce premature aging,
while pharmacological activation of Nrf2 can increase cell
lifespan, suggesting that inhibition of Nrf2 signaling in the
context of oxidative stress is able to facilitate premature
cell aging [50]. Currently, numerous comprehensive studies have demonstrated that the role of the Nrf2 transcription factor plays an essential role in maintaining cartilage
homeostasis and regulating redox balance in OA. Antioxidant and detoxification enzymes such as heme oxygenase
1 (HO-1), sulfiredoxin (Srx), thioredoxin reductase (TrxR),
peroxiredoxins (Prxs), catalase, SODs, glutathione peroxidase (GPx), and NADPH:quinone oxidoreductase 1
(NQO1) can be significantly regulated and induced by
Nrf2. Therefore, maintaining the stability of Nrf2 physiological function exerts an essential effect in maintaining
cellular redox homeostasis [51]. Recent studies have confirmed that activation of Nrf2 can reduce IL-1β-induced
ROS production in chondrocytes, further suggesting that
Nrf2 is an important determinant of antioxidant response
[52, 53]. According to the study by Wang et al., the level
of Nrf2 protein was decreased in human OA chondrocytes
compared with healthy chondrocytes [54]. Interestingly,
other studies have confirmed that Nrf2 protein levels are
increased in OA cartilage and synovium compared to
normal individuals [52, 53]. Furthermore, Khan et al. proposed that Nrf2 gene expression is dramatically elevated
in severely injured OA cartilage compared to uninjured
samples from the same OA joint [53].
Recently, Nrf2 has been shown to play an important role
in maintaining cartilage homeostasis in vivo. Notably,
Wruck et al. have found and confirmed that in the mouse
model of rheumatoid arthritis, Nrf2 knockout mice exhibited higher levels of oxidative stress and more severe
articular cartilage damage, compared to wild-type mice
[55]. Furthermore, according to Cai et al., Nrf2 knockout
mice exhibited a more severe OA phenotype compared to
wild-type mice in both post-traumatic OA models and inflammatory OA models [56]. Meanwhile, histone deacetylation inhibitors (TSAs) have been shown to ameliorate
the progression of OA in the above two mouse models via
TSA-induced acetylation and Nrf2 activation pathways. In
chondrocytes, downstream antioxidant mediators (such as
HO-1) are activated by acetylation-induced Nrf2 and lead
to the upregulation of its expression, which is related to
the reduction of matrix metalloproteinase expression [56].
HO-1 is considered to be an important downstream target
of Nrf2, and recent evidence has indicated that knockout
of Bach1, which is a transcriptional suppressor factor of
HO-1, can alleviate the severity of age-related OA and
surgically induced OA in mice [57]. In vitro studies have
shown that articular chondrocytes of Bach1 knockout
mice have higher levels of SOD2 protein, which is a key
superoxide detoxifying antioxidant. The level of SOD2
was found to decrease in Bach1 knockout cells after HO-1
gene expression was silenced, indicating that the expression of SOD2 is dependent on HO-1. In addition, Takada
et al. demonstrated that TBHP-induced chondrocyte apoptosis was enhanced when HO-1 expression was silenced,
further demonstrating that HO-1 has an essential effect in
the dynamic redox balance of cartilage (Figure 1) [57].
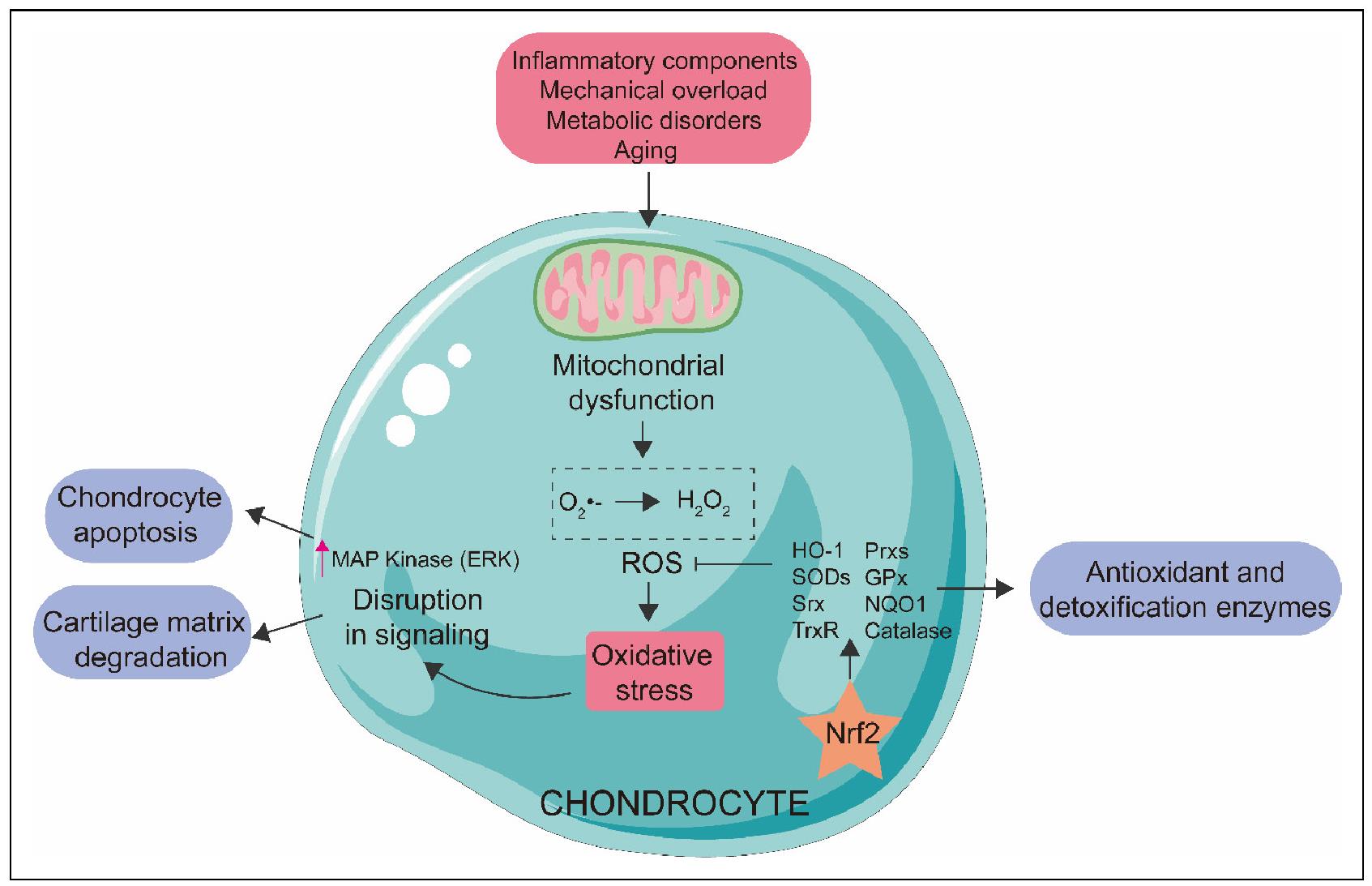
Figure 1. Nrf2 modulates oxidative stress in OA. Various negative stimuli, including inflammatory components, mechanical overload, metabolic disorders and aging, impair the function of the electron transport chain, leading to mitochondrial dysfunction. Then, the ROS system is activated, inducing oxidative stress, which affects cell signaling through the MAP kinase pathway, ultimately contributing to chondrocyte apoptosis and cartilage matrix degradation. Nrf2 is able to modulate antioxidant and detoxification enzymes such as heme oxygenase 1 (HO-1), sulfiredoxin (Srx), thioredoxin reductase (TrxR), peroxiredoxins (Prxs), catalase, SODs, glutathione peroxidase (GPx), and NADPH: quinone oxidoreductase 1 (NQO1), which inhibits the ROS system and thus reduces oxidative stress, further maintaining chondrocyte redox homeostasis and preventing progression of OA.
Based on accumulating evidence, various types of compounds with antioxidant properties, such as licochalcone A, pterostilbene, resveratroland, wogonin, and 6-gingero, etc., have been identified to play anti-inflammatory as well as cartilage protective roles in the joint of OA via activation of Nrf2 signaling pathways (Table 1). In light of these findings, it is reasonable to speculate that Nrf2 signaling pathways contribute to the maintenance of redox balance and modulation of chondrocyte homeostasis in the process of OA, but the exact signaling pathways responsible for these effects are not fully understood at present. According to the results of Khan et al., Nrf2 can modulate IL-1β- induced ROS production by stimulating the ERK MAP kinase pathway, thereby promoting the anti-apoptotic effect, which further elucidates that the regulation of Nrf2- controlled signaling pathways may have an important effect in attenuating oxidation and apoptosis of human OA chondrocytes [53]. Additionally, the in-depth study of Nrf2 has not yet identified any clinical drugs targeting OA, but a variety of new clinical drugs have been developed for the treatment of other diseases. For example, based on a randomized, double-blind, placebo-controlled phase 3 clinical trial (CARDINAL), the Nrf2 activator bardoxolone methyl was approved by the U.S. Food and Drug Administration (FDA) for the treatment of chronic kidney disease (CKD) caused by Alport syndrome [58]. Another Nrf2 activator, dimethyl fumarate, can induce upregulation of antioxidant gene expression, and is therefore also approved by the FDA and the European Medicines Agency as a first‐line therapy for adult patients with relapsing‐remitting MS (RMSS) [59]. However, activation of Nrf2 has been shown to play a double-edged role in cancer. On the one hand, Nrf2 can prevent cancer progression caused by oxidative stress; on the other hand, specific activation of Nrf2 in various cancers can promote cancer cell proliferation as well as induce chemo- and radioresistance of cancer cells [60]. Therefore, potential therapeutic strategies that precisely target Nrf2-regulated pathways may be of clinical value in ameliorating the progression of OA, and much remains to be done.
Table 1
Treatment with bioactive agents with antioxidant activity for
OA by targeting Nrf2.
Conclusion and future perspectives
Accumulating evidence suggests that OA is not only a chronic injury disease limited to the joints, but also a comprehensive and degenerative disease involving multiple systems. There is increasing evidence that oxidative stress plays a dramatic role in age-related changes in articular cartilage, disrupting cartilage homeostasis and contributing to the development of OA. Aging, inflammation, and mechanical loading are capable of inducing oxidative stress and promoting ROS production that damages proteins and DNA, resulting in mitochondrial dysfunction, disruption of cell signaling, and alterations in epigenetic gene expression. Therefore, high levels of ROS signaling pathways and altered Nrf2 activity may facilitate chondrocyte apoptosis along with cartilage degradation and induce chondrocyte hypertrophy and subchondral bone dysfunction. In addition, decreased Nrf2 activity may be the result of a failure in its homeostatic post-translational regulation and/or altered epigenetic and transcriptional regulatory mechanisms. A large body of experimental evidence indicates that Nrf2 plays a central and complex role in bone integrity, and many functions remain to be elucidated. Treatment methods that increase Nrf2 activity may counteract oxidative stress in OA, thereby effectively limiting cartilage degradation and bone resorption, while restoring the dynamic balance of Nrf2 may induce normalization of bone resorption and remodeling. Thus, the study of the effect of chondrocytes and Nrf2 in OA would be a promising research area for the development of a potential therapeutic strategy for the treatment of OA.
Declarations
Availability of data and materials
Not applicable.
Financial support and sponsorship
None.
Conflicts of interest
The authors report no conflicts of interest.
Ethical approval and informed consent statement
Not applicable.
Consent for publication
Not applicable.
References
1. Long H, Liu Q, Yin H, Wang K, Diao N, Zhang Y, et al. Prevalence Trends of Site-Specific Osteoarthritis From 1990 to 2019: Findings From the Global Burden of Disease Study 2019. Arthritis Rheumatol, 2022, 74(7): 1172- 1183. [Crossref]
2. Martel-Pelletier J, Barr AJ, Cicuttini FM, Conaghan PG, Cooper C, Goldring MB, et al. Osteoarthritis. Nat Rev Dis Primers, 2016, 2: 16072. [Crossref]
3. Blagojevic M, Jinks C, Jeffery A, & Jordan KP. Risk factors for onset of osteoarthritis of the knee in older adults: a systematic review and meta-analysis. Osteoarthritis Cartilage, 2010, 18(1): 24-33. [Crossref]
4. Price JS, Waters JG, Darrah C, Pennington C, Edwards DR, Donell ST, et al. The role of chondrocyte senescence in osteoarthritis. Aging Cell, 2002, 1(1): 57-65. [Crossref]
5. Marchev AS, Dimitrova PA, Burns AJ, Kostov RV, DinkovaKostova AT, & Georgiev MI. Oxidative stress and chronic inflammation in osteoarthritis: can NRF2 counteract these partners in crime? Ann N Y Acad Sci, 2017, 1401(1): 114-135. [Crossref]
6. Cheung EC, & Vousden KH. The role of ROS in tumour development and progression. Nat Rev Cancer, 2022, 22(5): 280-297. [Crossref]
7. Mittler R. ROS Are Good. Trends Plant Sci, 2017, 22(1): 11-19. [Crossref]
8. Gavriilidis C, Miwa S, von Zglinicki T, Taylor RW, & Young DA. Mitochondrial dysfunction in osteoarthritis is associated with down-regulation of superoxide dismutase 2. Arthritis Rheum, 2013, 65(2): 378-387. [Crossref]
9. He F, Antonucci L, & Karin M. NRF2 as a regulator of cell metabolism and inflammation in cancer. Carcinogenesis, 2020, 41(4): 405-416. [Crossref]
10. Kensler TW, Wakabayashi N, & Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2- ARE pathway. Annu Rev Pharmacol Toxicol, 2007, 47: 89- 116. [Crossref]
11. Ludtmann MH, Angelova PR, Zhang Y, Abramov AY, & Dinkova-Kostova AT. Nrf2 affects the efficiency of mitochondrial fatty acid oxidation. Biochem J, 2014, 457(3): 415-424. [Crossref]
12. Mitsuishi Y, Taguchi K, Kawatani Y, Shibata T, Nukiwa T, Aburatani H, et al. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell, 2012, 22(1): 66-79. [Crossref]
13. McMahon M, Thomas N, Itoh K, Yamamoto M, & Hayes JD. Redox-regulated turnover of Nrf2 is determined by at least two separate protein domains, the redox-sensitive Neh2 degron and the redox-insensitive Neh6 degron. J Biol Chem, 2004, 279(30): 31556-31567. [Crossref]
14. Chang M, Wilson CJ, Karunatilleke NC, Moselhy MH, Karttunen M, & Choy WY. Exploring the Conformational Landscape of the Neh4 and Neh5 Domains of Nrf2 Using Two Different Force Fields and Circular Dichroism. J Chem Theory Comput, 2021, 17(5): 3145-3156. [Crossref]
15. Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun, 1997, 236(2): 313-322. [Crossref]
16. Katsuoka F, Motohashi H, Ishii T, Aburatani H, Engel JD, & Yamamoto M. Genetic evidence that small maf proteins are essential for the activation of antioxidant response element-dependent genes. Mol Cell Biol, 2005, 25(18): 8044-8051. [Crossref]
17. Plafker KS, Nguyen L, Barneche M, Mirza S, Crawford D, & Plafker SM. The ubiquitin-conjugating enzyme UbcM2 can regulate the stability and activity of the antioxidant transcription factor Nrf2. J Biol Chem, 2010, 285(30): 23064-23074. [Crossref]
18. Wang H, Liu K, Geng M, Gao P, Wu X, Hai Y, et al. RXRα inhibits the NRF2-ARE signaling pathway through a direct interaction with the Neh7 domain of NRF2. Cancer Res, 2013, 73(10): 3097-3108. [Crossref]
19. Hinoi E, Takarada T, Fujimori S, Wang L, Iemata M, Uno K, et al. Nuclear factor E2 p45-related factor 2 negatively regulates chondrogenesis. Bone, 2007, 40(2): 337-344. [Crossref]
20. Solomon LA, Bérubé NG, & Beier F. Transcriptional regulators of chondrocyte hypertrophy. Birth Defects Res C Embryo Today, 2008, 84(2): 123-130. [Crossref]
21. Liu M, Liu S, Zhang Q, Fang Y, Yu Y, Zhu L, et al. Curculigoside attenuates oxidative stress and osteoclastogenesis via modulating Nrf2/NF-κB signaling pathway in RAW264.7 cells. J Ethnopharmacol, 2021, 275: 114129. [Crossref]
22. Hunter DJ, & Bierma-Zeinstra S. Osteoarthritis. Lancet, 2019, 393(10182): 1745-1759. [Crossref]
23. Prieto-Alhambra D, Judge A, Javaid MK, Cooper C, DiezPerez A, & Arden NK. Incidence and risk factors for clinically diagnosed knee, hip and hand osteoarthritis: influences of age, gender and osteoarthritis affecting other joints. Ann Rheum Dis, 2014, 73(9): 1659-1664. [Crossref]
24. Hunter DJ, Schofield D, & Callander E. The individual and socioeconomic impact of osteoarthritis. Nat Rev Rheumatol, 2014, 10(7): 437-441. [Crossref]
25. Fu K, Robbins SR, & McDougall JJ. Osteoarthritis: the genesis of pain. Rheumatology (Oxford), 2018, 57(suppl_4): iv43-iv50. [Crossref]
26. Hu W, Chen Y, Dou C, & Dong S. Microenvironment in subchondral bone: predominant regulator for the treatment of osteoarthritis. Ann Rheum Dis, 2021, 80(4): 413- 422. [Crossref]
27. Scanzello CR. Role of low-grade inflammation in osteoarthritis. Curr Opin Rheumatol, 2017, 29(1): 79-85. [Crossref]
28. Bierma-Zeinstra SM, & van Middelkoop M. Osteoarthritis: In search of phenotypes. Nat Rev Rheumatol, 2017, 13(12): 705-706. [Crossref]
29. Courties A, Sellam J, & Berenbaum F. Metabolic syndrome-associated osteoarthritis. Curr Opin Rheumatol, 2017, 29(2): 214-222. [Crossref]
30. Loeser RF, Goldring SR, Scanzello CR, & Goldring MB. Osteoarthritis: a disease of the joint as an organ. Arthritis Rheum, 2012, 64(6): 1697-1707. [Crossref]
31. Liu-Bryan R, & Terkeltaub R. Emerging regulators of the inflammatory process in osteoarthritis. Nat Rev Rheumatol, 2015, 11(1): 35-44. [Crossref]
32. Moon PM, & Beier F. Novel Insights into Osteoarthritis Joint Pathology from Studies in Mice. Curr Rheumatol Rep, 2015, 17(8): 50-61. [Crossref]
33. Sellam J, & Berenbaum F. The role of synovitis in pathophysiology and clinical symptoms of osteoarthritis. Nat Rev Rheumatol, 2010, 6(11): 625-635. [Crossref]
34. Sandell LJ. Etiology of osteoarthritis: genetics and synovial joint development. Nat Rev Rheumatol, 2012, 8(2): 77-89. [Crossref]
35. Glyn-Jones S, Palmer AJ, Agricola R, Price AJ, Vincent TL, Weinans H, et al. Osteoarthritis. Lancet, 2015, 386(9991): 376-387. [Crossref]
36. Venkataraman K, Khurana S, & Tai TC. Oxidative stress in aging--matters of the heart and mind. Int J Mol Sci, 2013, 14(9): 17897-17925. [Crossref]
37. Jones DP. Redox theory of aging. Redox Biol, 2015, 5: 71- 79. [Crossref]
38. Hui W, Young DA, Rowan AD, Xu X, Cawston TE, & Proctor CJ. Oxidative changes and signalling pathways are pivotal in initiating age-related changes in articular cartilage. Ann Rheum Dis, 2016, 75(2): 449-458. [Crossref]
39. Loeser RF. The Role of Aging in the Development of Osteoarthritis. Trans Am Clin Climatol Assoc, 2017, 128: 44- 54.
40. Nathan C, & Cunningham-Bussel A. Beyond oxidative stress: an immunologist’s guide to reactive oxygen species. Nat Rev Immunol, 2013, 13(5): 349-361. [Crossref]
41. Paulsen CE, & Carroll KS. Orchestrating redox signaling networks through regulatory cysteine switches. ACS Chem Biol, 2010, 5(1): 47-62. [Crossref]
42. Li YS, Xiao WF, & Luo W. Cellular aging towards osteoarthritis. Mech Ageing Dev, 2017, 162: 80-84. [Crossref]
43. Chen K, Craige SE, & Keaney JF, Jr. Downstream targets and intracellular compartmentalization in Nox signaling. Antioxid Redox Signal, 2009, 11(10): 2467-2480. [Crossref]
44. Wu RF, Xu YC, Ma Z, Nwariaku FE, Sarosi GA, Jr., & Terada LS. Subcellular targeting of oxidants during endothelial cell migration. J Cell Biol, 2005, 171(5): 893-904. [Crossref]
45. De la Fuente M, & Miquel J. An update of the oxidationinflammation theory of aging: the involvement of the immune system in oxi-inflamm-aging. Curr Pharm Des, 2009, 15(26): 3003-3026. [Crossref]
46. Minguzzi M, Cetrullo S, D’Adamo S, Silvestri Y, Flamigni F, & Borzì RM. Emerging Players at the Intersection of Chondrocyte Loss of Maturational Arrest, Oxidative Stress, Senescence and Low-Grade Inflammation in Osteoarthritis. Oxid Med Cell Longev, 2018, 2018: 3075293. [Crossref]
47. Bolduc JA, Collins JA, & Loeser RF. Reactive oxygen species, aging and articular cartilage homeostasis. Free Radic Biol Med, 2019, 132: 73-82. [Crossref]
48. Yudoh K, Nguyen v T, Nakamura H, Hongo-Masuko K, Kato T, & Nishioka K. Potential involvement of oxidative stress in cartilage senescence and development of osteoarthritis: oxidative stress induces chondrocyte telomere instability and downregulation of chondrocyte function. Arthritis Res Ther, 2005, 7(2): R380-391. [Crossref]
49. Brandl A, Hartmann A, Bechmann V, Graf B, Nerlich M, & Angele P. Oxidative stress induces senescence in chondrocytes. J Orthop Res, 2011, 29(7): 1114-1120. [Crossref]
50. Zinovkin RA, Kondratenko ND, & Zinovkina LA. Does Nrf2 Play a Role of a Master Regulator of Mammalian Aging? Biochemistry (Mosc), 2022, 87(12): 1465-1476. [Crossref]
51. Tebay LE, Robertson H, Durant ST, Vitale SR, Penning TM, Dinkova-Kostova AT, et al. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radic Biol Med, 2015, 88(Pt B): 108-146. [Crossref]
52. Vaamonde-Garcia C, Courties A, Pigenet A, Laiguillon MC, Sautet A, Houard X, et al. The nuclear factor-erythroid 2-related factor/heme oxygenase-1 axis is critical for the inflammatory features of type 2 diabetes-associated osteoarthritis. J Biol Chem, 2017, 292(35): 14505-14515. [Crossref]
53. Khan NM, Ahmad I, & Haqqi TM. Nrf2/ARE pathway attenuates oxidative and apoptotic response in human osteoarthritis chondrocytes by activating ERK1/2/ELK1- P70S6K-P90RSK signaling axis. Free Radic Biol Med, 2018, 116: 159-171. [Crossref]
54. Wang Y, Zhao X, Lotz M, Terkeltaub R, & Liu-Bryan R. Mitochondrial biogenesis is impaired in osteoarthritis chondrocytes but reversible via peroxisome proliferatoractivated receptor γ coactivator 1α. Arthritis Rheumatol, 2015, 67(8): 2141-2153. [Crossref]
55. Wruck CJ, Fragoulis A, Gurzynski A, Brandenburg LO, Kan YW, Chan K, et al. Role of oxidative stress in rheumatoid arthritis: insights from the Nrf2-knockout mice. Ann Rheum Dis, 2011, 70(5): 844-850. [Crossref]
56. Cai D, Yin S, Yang J, Jiang Q, & Cao W. Histone deacetylase inhibition activates Nrf2 and protects against osteoarthritis. Arthritis Res Ther, 2015, 17: 269-280. [Crossref]
57. Takada T, Miyaki S, Ishitobi H, Hirai Y, Nakasa T, Igarashi K, et al. Bach1 deficiency reduces severity of osteoarthritis through upregulation of heme oxygenase-1. Arthritis Res Ther, 2015, 17: 285-296. [Crossref]
58. Warady BA, Pergola PE, Agarwal R, Andreoli S, Appel GB, Bangalore S, et al. Effects of Bardoxolone Methyl in Alport Syndrome. Clin J Am Soc Nephrol, 2022, 17(12): 1763-1774. [Crossref]
59. Xu Z, Zhang F, Sun F, Gu K, Dong S, & He D. Dimethyl fumarate for multiple sclerosis. Cochrane Database Syst Rev, 2015, (4): Cd011076. [Crossref]
60. Qin JJ, Cheng XD, Zhang J, & Zhang WD. Dual roles and therapeutic potential of Keap1-Nrf2 pathway in pancreatic cancer: a systematic review. Cell Commun Signal, 2019, 17(1): 121-136. [Crossref]